
Moleküler Orbital Teorisi: Kuantum Mekaniği ile Kimyasal Bağların Keşfi
- Emre Can Buluz

- 3 Nis 2025
- 4 dakikada okunur
Güncelleme tarihi: 10 Haz 2025
Moleküler orbital teorisi, atomların bir araya gelerek molekülleri oluştururken nasıl bağlandığını ve elektronların bu bağlar içinde nasıl dağıldığını açıklayan temel bir kuantum mekaniği yaklaşımıdır. Bu teori, atomik orbitallerin birleşerek moleküler orbitalleri oluşturduğunu ve bu orbitallerin, elektronların olasılık yoğunluklarını belirlediğini öne sürer. Moleküler bağların gücünü, manyetik özelliklerini ve elektronik geçişlerini anlamada kritik bir rol oynayan moleküler orbital teorisi, özellikle kimyasal reaksiyonların mekanizmalarını ve spektroskopik özellikleri açıklamada yaygın olarak kullanılmaktadır. Bu yazıda, moleküler orbital teorisinin temel prensipleri, bağ oluşumuna etkisi ve hesaplamalı kimyada nasıl uygulandığı ele alınacaktır.
Moleküler orbital teorisi kovalent bağları, bağlanan atomların atomik orbitallerinin etkileşimi sonucu oluşan ve tüm molekülle ilişkili olan moleküler orbitaller açısından açıklar. Moleküler orbital ile atomik orbital arasındaki fark, atomik orbitalin yalnızca tek bir atomla ilişkili olmasıdır, oysa moleküler orbital tüm moleküle yayılmıştır. Moleküler orbital (MO) teorisine göre, iki hidrojen atomunun 1s orbitallerinin örtüşmesi, bir bağlayıcı (bonding) moleküler orbital ve bir karşıt bağlayıcı (antibonding) moleküler orbital olmak üzere iki moleküler orbitalin oluşmasına neden olur. Bağlayıcı moleküler orbital, kendisini oluşturan atomik orbitallerden daha düşük enerjiye ve daha yüksek kararlılığa sahiptir. Karşıt bağlayıcı moleküler orbital ise, atomik orbitallerden daha yüksek enerjiye ve daha düşük kararlılığa sahiptir. "Bağlayıcı" ve "karşıt bağlayıcı" terimlerinin de gösterdiği gibi elektronların bağlayıcı moleküler orbitale yerleşmesi kararlı bir kovalent bağın oluşmasına neden olurken elektronların karşıt bağlayıcı moleküler orbitale yerleşmesi kararsız bir bağın oluşmasına yol açmaktadır.
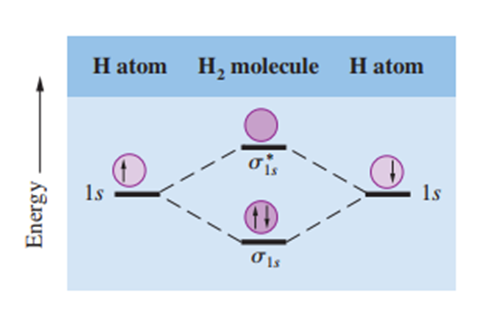
Bağlayıcı moleküler orbitallerde elektron yoğunluğu, bağlanan atomların çekirdekleri arasında en yüksek seviyededir. Karşıt bağlayıcı moleküler orbitallerde ise elektron yoğunluğu, çekirdekler arasında sıfıra düşer. Bu farkı anlamak için orbitallerdeki elektronların dalga özelliklerine sahip olduğunu hatırlamak gerekir. Dalgalara özgü bir özellik, aynı türdeki dalgaların birbirleriyle etkileşerek ya genliklerinin artmasına ya da azalmasına neden olmasıdır. Bağlayıcı moleküler orbitallerin oluşumu, yapıcı girişime karşılık gelir (genlik artışı, iki çekirdek arasındaki elektron yoğunluğunun artmasına benzer). Karşıt bağlayıcı moleküler orbitallerin oluşumu ise yıkıcı girişime karşılık gelir (genlik azalması, iki çekirdek arasındaki elektron yoğunluğunun azalmasına benzer) (1). Bu nedenle, H₂ molekülündeki iki 1s orbitalinin yapıcı ve yıkıcı etkileşimleri, bir sigma bağlayıcı moleküler orbitalin (σ1s) ve bir sigma karşıt bağlayıcı moleküler orbitalin (σ*1s) oluşmasına neden olur (Şekil 1.).
Sigma Moleküler Orbitaller
Bir sigma moleküler orbitalinde (bağlayıcı veya karşıt bağlayıcı) elektron yoğunluğu, bağ yapan atomların iki çekirdeği arasındaki bir çizgi boyunca simetrik olarak yoğunlaşır. Bir sigma moleküler orbitalindeki iki elektron bir sigma bağı oluşturur. Tek bir kovalent bağ (örneğin, H–H veya F–F) neredeyse her zaman bir sigma bağıdır.
H₂ molekülünün oluşumu sırasında üretilen orbitallerin göreli enerji seviyelerini ve iki 1s orbitalinin yapıcı ve yıkıcı etkileşimlerini gösteren moleküler orbital enerji seviyesi diyagramı görülmektedir (Şekil 1.). Karşıt bağlayıcı moleküler orbitalinde, çekirdekler arasında elektron yoğunluğunun sıfır olduğu bir düğüm (node) bulunur. Bu durumda, çekirdekler birbirlerinin pozitif yükleri tarafından itilir ve bir arada tutulmazlar. Karşıt bağlayıcı moleküler orbitaldeki elektronlar, izole atomlardaki elektronlara kıyasla daha yüksek enerjiye (ve daha düşük stabiliteye) sahiptir. Öte yandan, bağlayıcı moleküler orbitalde bulunan elektronlar, izole atomlardaki elektronlara kıyasla daha düşük enerjiye (ve dolayısıyla daha yüksek stabiliteye) sahiptir (1).
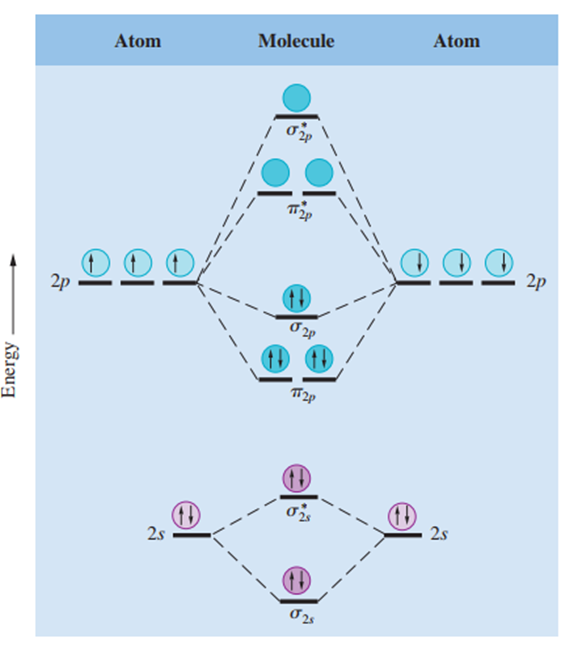
Pi Moleküler Orbitaller
Bir pi moleküler orbitalinde (bağlayıcı veya karşıt bağlayıcı) elektron yoğunluğu, bağ yapan atomların iki çekirdeğini birleştiren çizginin üstünde ve altında yoğunlaşır. Bir pi moleküler orbitalindeki iki elektron bir pi bağı oluşturur. Çift bağlar neredeyse her zaman bir sigma bağı ve bir pi bağından oluşurken, üçlü bağlar her zaman bir sigma bağı ve iki pi bağı içermektedir.
Moleküler Orbitallerin Kimyasal Reaksiyonlardaki Yeri
Moleküler orbitaller (MO'lar), kimyasal reaksiyonların mekanizmasını anlamada kritik bir rol oynamaktadır. Bir moleküldeki elektronların dağılımı, moleküler orbital teorisi kullanılarak incelendiğinde, reaksiyonun nasıl ilerleyeceği hakkında önemli ipuçları elde edilir. Özellikle, en yüksek dolu moleküler orbital (HOMO) ve en düşük boş moleküler orbital (LUMO) etkileşimleri, reaksiyonların gerçekleşme olasılığını belirleyen temel faktörlerdir. HOMO, bir molekülün nükleofilik özelliklerini, LUMO ise elektrofilik özelliklerini belirler. Sınır Moleküler Orbital Teori (Frontier Molecular Orbital Theory (FMOT)) olarak bilinen bu yaklaşım, Diels-Alder reaksiyonları, elektrofilik aromatik sübstitüsyonlar ve organokataliz gibi birçok organik kimyasal reaksiyonun mekanizmasını açıklamada yaygın olarak kullanılmaktadır. Ayrıca, kuantum kimyası teknikleri ile yapılan yoğunluk fonksiyonel teorisi (DFT) hesaplamaları sayesinde, reaktiflerin ve ürünlerin orbital enerjileri ile geçiş durumları ayrıntılı olarak incelenebilmektedir. Bu tür hesaplamalar, ilaç tasarımından malzeme bilimlerine kadar geniş bir alanda reaksiyon verimliliğini ve mekanizmalarını optimize etmek için kullanılır (3).
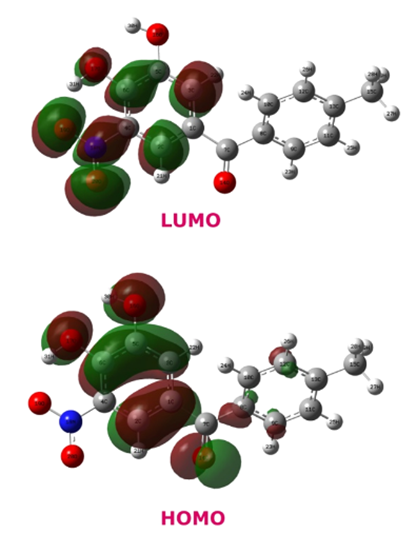
Ayrıca moleküler orbitaller, kimyasal reaksiyonların geçiş hallerinin incelenmesinde de kullanılır. Kuantum kimyası teknikleri, özellikle Yoğunluk Fonksiyonel Teorisi (DFT) ve Hartree-Fock gibi yöntemler, reaksiyon koordinatlarını modelleyerek enerji bariyerlerini ve kararsız ara ürünlerin yapılarını anlamamıza yardımcı olmaktadır. Örneğin, perisiklik reaksiyonlar (Diels-Alder, elektrosiklizasyon) ve radikalik süreçler, moleküler orbital simetrisi kullanılarak tahmin edilebilir (Woodward-Hoffmann Kuralları). Biyolojik sistemlerde de moleküler orbitallerin etkisi büyüktür. Enzim katalizli reaksiyonlarda, substrat ve enzim aktif bölgesi arasındaki HOMO-LUMO etkileşimleri, bağ yapma ve kırılma süreçlerini yönlendirir. Aynı zamanda fotokimya ve kuantum biyoloji alanlarında, ışık ile uyarılmış elektronların orbital değişimleri sayesinde biyolojik süreçler (örneğin, fotosentez ve görme mekanizması) açıklanabilir. Bu teorik kavramlar, ilaç tasarımı, kataliz, nanoteknoloji ve malzeme bilimi gibi disiplinlerde pratik uygulamalar bulur. Moleküler orbital hesaplamaları, yeni ilaç moleküllerinin biyomoleküllerle nasıl etkileşeceğini anlamak için kullanılırken, organik elektronikte π-konjuge sistemlerin tasarımında önemli bir rehber olmaktadır. Sonuç olarak, moleküler orbitaller kimyasal reaksiyonların temel mekanizmalarını açıklayan güçlü bir araç olup modern kimya ve biyokimyanın birçok alanında geniş uygulama alanına sahiptir (5,6).
Referanslar
1. Raymond Chang, Jason Overby. (2011). General Chemistry: The Essential Concepts. McGraw-Hill.
2. Darrell Ebbing, Steven D. Gammon. (2007). General Chemistry: Media Enhanced Edition. Cengage Learning.
3. Fukui, K. (1982). Role of frontier orbitals in chemical reactions. science, 218(4574), 747-754.
4. Isravel, A. D., Jeyaraj, J. K., Thangasamy, S., & John, W. J. (2021). DFT, NBO, HOMO-LUMO, NCI, stability, Fukui function and hole–Electron analyses of tolcapone. Computational and Theoretical Chemistry, 1202, 113296.
5. Woodward, R. B., & Hoffmann, R. (1969). The Conservation of Orbital Symmetry. Angewandte Chemie International Edition, 8(11), 781-853. https://doi.org/10.1002/anie.196907811
6. Parr, R. G., & Yang, W. (1989). Density-Functional Theory of Atoms and Molecules. Oxford University Press.



Yorumlar