
Moleküler Modellemenin Tarihçesi ve Temel Kavramlar
- Ege Altun

- 3 Nis 2025
- 5 dakikada okunur
Güncelleme tarihi: 10 Haz 2025
Moleküler Modelleme Nedir?
Moleküler modelleme, atomların ve moleküllerin fiziksel ve kimyasal özelliklerini anlamak, tahmin etmek, davranışlarını analiz etmek veya görselleştirmek için bilgisayar destekli yöntemlerin kullanıldığı bir tekniktir. Özellikle ilaç tasarımı, malzeme bilimi ve biyoteknoloji gibi alanlarda oldukça büyük bir öneme sahiptir.
Moleküler Modellemenin Tarihsel Gelişimi
Moleküler modelleme, temellerini 1920'li ve 30'lu yıllarda kuantum mekaniğinin gelişimi ile atmıştır. Erwin Schrödinger ve Werner Heisenberg gibi bilim insanlarının çalışmaları, atom ve moleküllerin davranışlarını anlamak için teorik bir çerçeve sunmuştur. Schrödinger denklemi, elektronların bir molekül içinde nasıl davrandığını tahmin ederek bağ yapılarının ve moleküler enerjilerin hesaplanmasını mümkün kılmıştır.
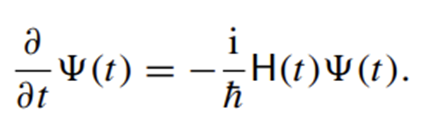
1940'larda Newton mekaniğini kullanarak molekül yapılarını analiz etmeye yönelik çalışmalar yapılmıştır. Bu dönemde, bağ uzunlukları ve açılarını hesaplamak için teorik modeller geliştirilmiş, sterik etkileşimler (atomların fiziki yakınlığı nedeniyle oluşan itme kuvvetleri) ve enerji minimumları (bir sistemin en kararlı ve düşük enerjili durumu) incelenmiştir. Moleküllerin yapısal değişimlerinin enerjisel etkilerini araştıran bu çalışmalar, modern moleküler modelleme tekniklerinin temelini oluşturmuştur (2).
1953 yılında, Monte Carlo yöntemleri ve Metropolis algoritması geliştirilerek modern simülasyon tekniklerinin temelleri atılmıştır (4). 1957'de Alder ve Wainwright, "Sert Küre (Hard Sphere)" modeli olarak bilinen ilk moleküler dinamik simülasyonu gerçekleştirmiştir (5). 1960'lı yıllarda ise bilgisayarlar, kuvvet alanlarının hesaplanmasında kullanılmaya başlanmış ve konformasyonel analizler için algoritmalar geliştirilmiştir. 1971'de Protein Data Bank (PDB) (6) kurulmuş ve 1977'de ilk protein dinamik simülasyonu gerçekleştirilmiştir (7). 1980'lerde ise kişisel bilgisayarların ve grafik arayüzlerin (GUI) yaygınlaşmasıyla moleküler modelleme daha erişilebilir hale gelmiştir. 1990'lı yıllardan itibaren, yüksek performanslı bilgisayarlar ve gelişmiş yazılımlar (örneğin GROMACS (8) ve AMBER (9)) sayesinde daha karmaşık moleküler sistemlerin modellenmesi mümkün olmuştur.
Moleküler Modelleme için Temel Kavramlar:
Atomlar ve Moleküller: Atomlar, moleküllerin yapı taşlarıdır. İki veya daha fazla atomun kimyasal bağlarla birleşmesiyle moleküller oluşur. Moleküllerin yapısal düzeni, kimyasal özelliklerini belirlemektedir.
Kimyasal Bağlar: Atomlar arasında oluşan ve molekülleri bir arada tutan bağlar, molekülün stabilitesini ve fonksiyonunu belirler. Modelleme süreçlerinde bu bağların kuvvetleri ve dinamikleri analiz edilmektedir.
Moleküler Geometri: Moleküler geometri, atomların bir molekül içinde uzaydaki düzenini tanımlar. Bağ açısı, bağ uzunluğu ve molekülün üç boyutlu şekli, kimyasal özellikleri doğrudan etkilemektedir.
Potansiyel Enerji Yüzeyi (PES): Potansiyel enerji yüzeyi, moleküler sistemin toplam enerjisinin, atomların farklı geometrik konumlarına bağlı olarak nasıl değiştiğini gösterir. PES, molekülün kararlı durumlarını ve geçiş durumlarını belirler. Bu bilgi, kimyasal reaksiyonların enerji bariyerlerini ve olası yollarını anlamak için kullanılmaktadır.
Kuvvet Alanları (Force Fields): Kuvvet alanları, moleküller arasındaki etkileşimleri hesaplamak için kullanılan matematiksel modellerdir. Kuvvet alanları, atomlar arasındaki bağ kuvvetlerini, açısal gerginlikleri ve van der Waals kuvvetlerini içerir. Yaygın kullanılan kuvvet alanları arasında AMBER (11), CHARMM (12) ve GROMOS (13) bulunur ve moleküler dinamik simülasyonlarının temelini oluşturmaktadırlar.
Moleküler Dinamik (MD) Simülasyonları: Moleküler dinamik simülasyonları, moleküllerin zamanla nasıl hareket ettiğini anlamak için yapılan simülasyonlardır. Atomlar arasındaki kuvvetler, Newton’un hareket yasaları kullanılarak molekülün dinamik davranışları tahmin edilir. Moleküler dinamik, zamanla moleküllerin stabilitesini ve esnekliğinin incelenmesini de sağlamaktadır.
Bilgisayarla Modellemenin Önemi
Bilgisayarla moleküler modelleme, moleküllerin atomik düzeydeki yapılarını ve etkileşimlerini anlamamızı sağlamaktadır. Özellikle karmaşık biyolojik sistemlerin modellenmesi, bilgisayar sayesinde hem daha hızlı hem de daha ekonomik bir şekilde gerçekleştirilebilir. Bilgisayar destekli modelleme, fiziksel deney seti oluşturmanın maliyetli ve zaman alıcı olduğu durumlarda alternatif bir çözüm sunarak süreçleri hızlandırır ve maliyetleri düşürür. Ayrıca, deneysel yöntemlerle gözlemlenmesi zor veya imkansız olan süreçler, bilgisayarla moleküler düzeyde yapılan simülasyonlarla anlaşılabilir. Çünkü bilgisayar, atomik düzeyde milyonlarca parçacığın etkileşimlerini simüle ederek laboratuvarlarda erişilemeyen detaylara ulaşmamıza olanak tanımaktadır. Bu yöntem, özellikle ilaç keşfi (14), protein yapı analizi (15), enzim mühendisliği (16), kimyasal reaksiyon dinamiği (17) vs. gibi birçok alanda kullanılmaktadır.
Referanslar
1. Frank Vewinger, Bruce W. Shore, Klaas Bergmann. (2010). Chapter 3 - Superpositions of Degenerate Quantum States: Preparation and Detection in Atomic Beams, Pages 113-172, https://doi.org/10.1016/S1049-250X(10)05808-8
2. Terrell L. Hill; On Steric Effects. J. Chem. Phys. 1 July 1946; 14 (7): 465. https://doi.org/10.1063/1.1724172
3. Tamar Schlick. (2010). Molecular modeling and simulation: an interdisciplinary guide 2nd edition. Springer.
4. Nicholas Metropolis, Arianna W. Rosenbluth, Marshall N. Rosenbluth, Augusta H. Teller, Edward Teller; Equation of State Calculations by Fast Computing Machines. J. Chem. Phys. 1 June 1953; 21 (6): 1087–1092. https://doi.org/10.1063/1.1699114
5. B. J. Alder, T. E. Wainwright; Phase Transition for a Hard Sphere System. J. Chem. Phys. 1 November 1957; 27 (5): 1208–1209. https://doi.org/10.1063/1.1743957
6. H.M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T.N. Bhat, H. Weissig, I.N. Shindyalov, P.E. Bourne, The Protein Data Bank. (2000). Nucleic Acids Research 28: 235-242 https://doi.org/10.1093/nar/28.1.235.
7. McCammon, J., Gelin, B. & Karplus, M. Dynamics of folded proteins. Nature 267, 585–590 (1977). https://doi.org/10.1038/267585a0
8. Van Der Spoel, D., Lindahl, E., Hess, B., Groenhof, G., Mark, A. E., & Berendsen, H. J. (2005). GROMACS: fast, flexible, and free. Journal of computational chemistry, 26(16), 1701–1718.
9. R. Salomon-Ferrer, D.A. Case, R.C. Walker. (2013) "An overview of the Amber biomolecular simulation package." WIREs Comput. Mol. Sci. 3, 198-210.
10. Marcus D Hanwell, Donald E Curtis, David C Lonie, Tim Vandermeersch, Eva Zurek and Geoffrey R Hutchison; “Avogadro: An advanced semantic chemical editor, visualization, and analysis platform” Journal of Cheminformatics 2012, 4:17.
11. Wang, J., Wolf, R. M., Caldwell, J. W., Kollman, P. A., & Case, D. A. (2004).Development and testing of a general amber force field. Journal ofComputational Chemistry, 25(9), 1157–1174. https://doi.org/10.1002/jcc.20035
12. Huang, J., & MacKerell, A. D., Jr (2013). CHARMM36 all-atom additive protein force field: validation based on comparison to NMR data. Journal of computational chemistry, 34(25), 2135–2145. https://doi.org/10.1002/jcc.23354
13. Reif, M. M.; Hü nenberger, P. H.; Oostenbrink, C. J. Chem. Theory Comput. 2012, 8, 3705−3723.
14. Akash, S., Bayıl, I., Hossain, M. S., Islam, M. R., Hosen, M. E., Mekonnen, A. B., Nafidi, H. A., Bin Jardan, Y. A., Bourhia, M., & Bin Emran, T. (2023). Novel computational and drug design strategies for inhibition of human papillomavirus-associated cervical cancer and DNA polymerase theta receptor by Apigenin derivatives. Scientific reports, 13(1), 16565. https://doi.org/10.1038/s41598-023-43175-x
15. Dai, L., & Zhou, Y. (2011). Characterizing the existing and potential structural space of proteins by large-scale multiple loop permutations. Journal of molecular biology, 408(3), 585–595. https://doi.org/10.1016/j.jmb.2011.02.056
16. Richter, F., Blomberg, R., Khare, S. D., Kiss, G., Kuzin, A. P., Smith, A. J., Gallaher, J., Pianowski, Z., Helgeson, R. C., Grjasnow, A., Xiao, R., Seetharaman, J., Su, M., Vorobiev, S., Lew, S., Forouhar, F., Kornhaber, G. J., Hunt, J. F., Montelione, G. T., Tong, L., … Baker, D. (2012). Computational design of catalytic dyads and oxyanion holes for ester hydrolysis. Journal of the American Chemical Society, 134(39), 16197–16206. https://doi.org/10.1021/ja3037367
17. Grimme S. (2019). Exploration of Chemical Compound, Conformer, and Reaction Space with Meta-Dynamics Simulations Based on Tight-Binding Quantum Chemical Calculations. Journal of chemical theory and computation, 15(5), 2847–2862. https://doi.org/10.1021/acs.jctc.9b00143



Yorumlar