
Kimyasal Evrenin Topografyası: Potansiyel Enerji Yüzeyleri
- İlayda Boyraz

- 11 May 2025
- 5 dakikada okunur
Güncelleme tarihi: 10 Haz 2025
Potansiyel Enerji Yüzeyi Nedir?
Potansiyel enerji yüzeyi (PES), bir moleküler sistemdeki tüm atomların uzaydaki konumlarına karşılık gelen toplam enerji değerlerini temsil eden çok boyutlu bir yüzeydir. Her bir noktası belirli bir atomik konfigürasyona karşılık gelmektedir. Bu yüzeyin biçimi, bir molekülün kararlılığını, geçiş durumlarını ve reaksiyon yollarını anlamamızı sağlar. Kimyasal reaksiyonlarda reaktanların ürünlere dönüşümü, PES üzerinde bir yol boyunca gerçekleşir; bu yüzden PES, kimyasal reaksiyonların mekanizmalarını çözmede kritik bir araçtır (1).
Örneğin hidrojen alışverişi içeren bir asit-baz reaksiyonunda, protonun bir molekülden diğerine geçişi, PES üzerinde bir geçiş durumu (transition state) aracılığıyla modellenebilir. Bu durum, moleküler dinamik simülasyonlarında atomların nasıl yer değiştirdiğini izleyebilmek açısından da büyük avantaj sağlamaktadır. Dolayısıyla PES sadece teorik bir kavram değil, doğrudan gözlemlenebilir moleküler hareketlerin ardındaki fiziksel altyapıdır (2).
Born-Oppenheimer Yaklaşımı ve Potansiyel Enerji Yüzeyinin Oluşturulması
Potansiyel enerji yüzeyinin (PES) oluşturulması, Born-Oppenheimer yaklaşımına dayanmaktadır (Şekil 1). Bu yaklaşım, elektronların çekirdeğe göre çok daha hafif ve hızlı hareket ettiğini varsayarak, sistemin toplam dalga fonksiyonunu elektronik ve nükleer hareketlere ayırır. Böylece her bir çekirdek konfigürasyonu için elektronlar ayrı ayrı çözülebilir. Bu çözüm, çekirdek koordinatlarına bağlı bir potansiyel enerji fonksiyonu verir bu da PES’in kendisidir (3).
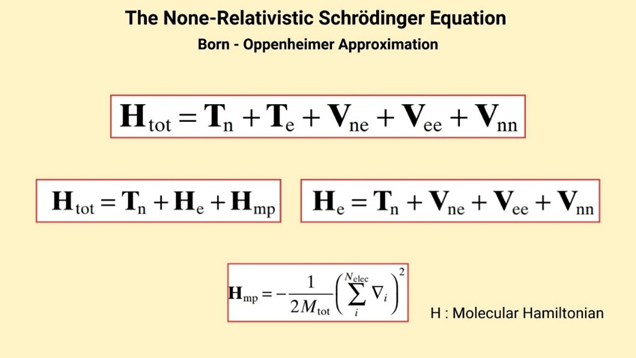
Bu ayrım, hesaplamalı kimyada büyük bir kolaylık sağlamıştır. Elektronik yapılar çözüldükten sonra, çekirdeklerin konumları optimize edilerek minimum enerji yapısı belirlenir. Eğer Born-Oppenheimer yaklaşımı kullanılmasaydı çok parçacıklı Schrödinger denklemini çözmek pratikte imkânsız hale gelebilirdi. Bu yüzden modern kuantum kimyasının temel taşı bu yaklaşımdır denebilir (4).
Potansiyel Enerji Yüzeyinin Yapısal Özellikleri
PES üzerinde minimum noktalar, sistemin kararlı yapılarını temsil eder; bu noktalar, enerji gradyanının sıfırlandığı ve Hessian matrisinin tüm özdeğerlerinin pozitif olduğu bölgelerdir. Bunlar gerçek moleküler yapıların karşılığıdır. Global minimum noktalar, sistemin en kararlı hâlini temsil etmektedir. Yerel minimumlar, geçici kararlı yapıların karşılığıdır. Öte yandan, saddle point (eğimli geçiş noktası) olarak bilinen noktalar, geçiş durumlarını (transition state) temsil etmektedir. Bu noktalarda enerji gradyanı sıfırdır ancak Hessian matrisinin en az bir özdeğeri negatiftir (5).
Kararlı Yapılar ve Minimum Noktalar
PES üzerinde minimum noktalar, sistemin kararlı yapılarını temsil eder. Bu noktalar, moleküler sistemin potansiyel enerjisinin yerel olarak en düşük olduğu konformasyonları ifade etmektedir. Bir kimyasal reaksiyon sürecinde, reaktan ve ürün yapıları bu minimumlarda yer alır (6).
Geçiş Durumları ve Enerji Bariyerleri
Geçiş durumları, iki minimumu birbirine bağlayan en düşük enerjili yol üzerindeki enerji tepe noktalarıdır. Bu noktalar, PES üzerinde "saddle point" (eyer noktası) olarak adlandırılmaktadır. Geçiş durumu, reaksiyon kinetiği açısından kritik öneme sahiptir; çünkü reaksiyonun aktivasyon enerjisi, reaktan minimumundan geçiş durumuna kadar olan enerji farkı ile belirlenmektedir (7). Bu nedenle bir kimyasal reaksiyonun mekanizmasını çözümlemek, potansiyel enerji yüzeyinde minimumları ve geçiş durumlarını bulmayı ve aralarındaki bağlantıları haritalamayı gerektirir. Bu işlem, minimum enerji yolu (minimum energy path (MEP)) veya içsel tepkime koordinatı (intrinsic reaction coordinate (IRC)) analizi ile yapılmaktadır (8).
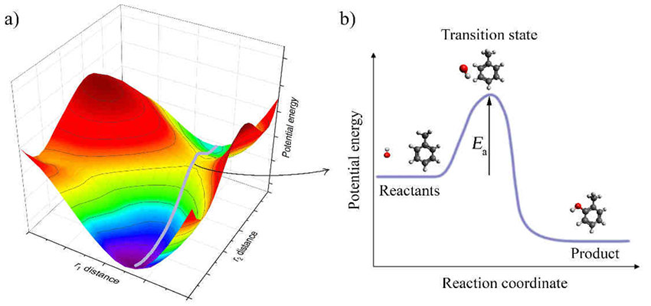
Bu hesaplamalar modern simülasyon yazılımları tarafından otomatik şekilde yapılmaktadır. Örneğin, Gaussian veya ORCA gibi yazılımlar hem gradyan hem de ikinci türev hesapları ile sistemin global minimuma ulaşmasını sağlar. Ayrıca moleküler dinamik simülasyonlarında kuvvet bilgileri, zaman içinde atomların nasıl hareket edeceğini belirleyen temel girdidir (12).
Kuvvetler ve Gradyan Hesapları
PES’in matematiksel türevleri ile sistemdeki atomik kuvvetler hesaplanabilir. Bir atom üzerindeki kuvvet, potansiyel enerjinin o koordinat yönündeki negatif gradyanıdır. Bu kuvvetlerin yardımıyla enerji minimizasyonları yapılmaktadır. Hessian matrisi (ikinci türev matrisi) ise sistemin titreşim modlarının ve geçiş durumu aramalarının yapılmasında kullanılır. Kuvvet ve gradyan bilgileri, özellikle geometri optimizasyonu ve geçiş durumu tahmini gibi algoritmalarda belirleyici rol oynamaktadır (10).
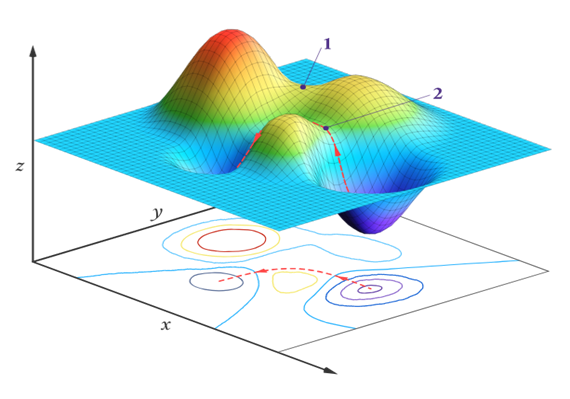
Bu hesaplamalar modern simülasyon yazılımları tarafından otomatik şekilde yapılmaktadır. Örneğin, Gaussian veya ORCA gibi yazılımlar hem gradyan hem de ikinci türev hesapları ile sistemin global minimuma ulaşmasını sağlar. Ayrıca moleküler dinamik simülasyonlarında kuvvet bilgileri, zaman içinde atomların nasıl hareket edeceğini belirleyen temel girdidir (12).
Titreşim Modları ve Spektroskopik Yorumlar
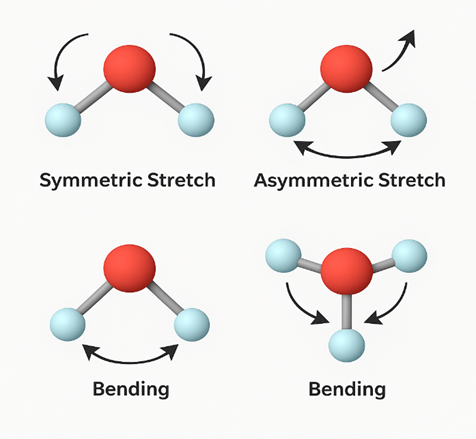
Bir molekülün PES üzerindeki denge konumu çevresinde yaptığı küçük titreşimler, harmonik osilatör yaklaşımı ile modellenebilmektedir. Bu yaklaşımda elde edilen normal modlar, Infrared (IR) ve Raman spektroskopisi gibi tekniklerle doğrudan gözlemlenebilir.
Her normal mod, molekülün kolektif bir hareketini temsil eder. Titreşim frekansları, Hessian matrisinin özdeğerleri ile bağlantılıdır ve PES’in lokal eğriliğine bağlıdır (13). Bu titreşim modları, özellikle bilinmeyen yapıların tayininde oldukça faydalıdır. Örneğin karbonil (C=O) grubunun karakteristik titreşimi ~1700 cm⁻¹ civarında olup, IR spektrumunda net bir pik verir. Bu tarz spektroskopik analizler, deneysel verilerin teorik hesaplamalarla doğrulanmasını sağlamaktadır (14).
Reaksiyon Enerji Profilleri ve Aktivasyon Enerjileri
Bir kimyasal reaksiyon sürecini tam olarak anlamak için yalnızca başlangıç ve bitiş durumlarını değil aradaki geçiş yollarını da bilmek gerekir. Potansiyel enerji yüzeyi, bu yolu açıkça tanımlamaktadır. Reaktanlar ve ürünler arasındaki minimum enerji yolu boyunca, sistem aktivasyon enerjisini aşarak geçiş durumundan geçer. Bu nedenle, bir reaksiyonun kinetiği ve termodinamiği doğrudan PES analizine dayanır. Bu yol boyunca geçilen en yüksek enerji noktası, geçiş durumudur (saddle point) ve bu noktadaki enerji ile reaktan minimumu arasındaki fark, aktivasyon enerjisidir. Reaksiyonun kinetiği bu enerji bariyerine bağlıdır. Özellikle radikal reaksiyonların veya fotokimyasal süreçlerin modellenmesinde bu profillerin nicel analizi büyük önem taşımaktadır (15).
Referanslar
Fukui, K. (1970). Formulation of the reaction coordinate. The Journal of Physical Chemistry, 74(23), 4161-4163.
Born, M., & Oppenheimer, R. (1927). Zur Quantentheorie der Molekeln. Annalen der Physik, 389(20), 457–484
Schlegel, H. B. (1982). Optimization of equilibrium geometries and transition structures. Journal of computational chemistry, 3(2), 214-218.
Jensen, F. (2017). Introduction to computational chemistry. John Wiley & Sons.
Eyring, H. (1935). The activated complex in chemical reactions. The Journal of chemical physics, 3(2), 107-115.
Fukui, K. (1981). The path of chemical reactions-the IRC approach. Accounts of chemical research, 14(12), 363-368.
Pople, J. A., & Beveridge, D. L. (1970). Molecular orbital theory. C0., NY.
Pulay, P. (2014). Analytical derivatives, forces, force constants, molecular geometries, and related response properties in electronic structure theory. Wiley Interdisciplinary Reviews: Computational Molecular Science, 4(3), 169-181. https://doi.org/10.1002/wcms.1171
Wacławek, S. (2021). Do we still need a laboratory to study advanced oxidation processes? A review of the modelling of radical reactions used for water treatment. Ecological Chemistry and Engineering, 28(1), 11-28. https://doi.org/10.2478/eces-2021-0002
Truhlar, D. G., & Garrett, B. C. (1980). Variational transition-state theory. Accounts of Chemical Research, 13(12), 440-448.
Libretexts. (2025, March 9). 30.10: The potential-energy surface can be calculated using quantum mechanics. Chemistry LibreTexts. https://chem.libretexts.org/Bookshelves/Physical_and_Theoretical_Chemistry_Textbook_Maps/Physical_Chemistry_%28LibreTexts%29/30%3A_Gas-Phase_Reaction_Dynamics/30.10%3A_The_Potential-Energy_Surface_Can_Be_Calculated_Using_Quantum_Mechanics
Mezei, M., & Beveridge, D. L. (1981). Theoretical studies of hydrogen bonding in liquid water and dilute aqueous solutions. The Journal of Chemical Physics, 74(1), 622-632.
Wilson, E. B., Decius, J. C., & Cross, P. C. (1980). Molecular vibrations: the theory of infrared and Raman vibrational spectra. Courier Corporation..
Bryce, D. L., Webster, F. X., Silverstein, R. M., & Kiemle, D. J. (2014). Spectrometric Identification of Organic Compounds.
Arrhenius, S. (1889). Über die Dissociationswärme und den Einfluss der Temperatur auf den Dissociationsgrad der Elektrolyte. Zeitschrift für physikalische Chemie, 4(1), 96-116.



Yorumlar