
Moleküler Dinamik Simülasyonlarında Zaman İntegrasyon Algoritmaları
- Emre Can Buluz

- 1 Mar
- 4 dakikada okunur
Moleküler dinamik (MD) simülasyonları, atomik düzeyde yapı–dinamik–fonksiyon ilişkilerini anlamada güçlü bir hesaplamalı araçtır ancak özellikle büyük biyomoleküler sistemlerde uzun zaman ölçeklerine ulaşmak ciddi hesaplama maliyetleri doğurmaktadır. Bu nedenle, klasik MD’nin temelini oluşturan hareket denklemlerinin sayısal entegrasyonundan kuvvet hesaplamalarının optimize edilmesine kadar pek çok “hız algoritması” geliştirilmiştir. Verlet tabanlı integratörlerin iyileştirilmiş versiyonları, çoklu zaman adımı yaklaşımları (ör. RESPA), kısıt algoritmaları (SHAKE/LINCS), Particle Mesh Ewald (PME) gibi hızlı elektrostatik yöntemler ve GPU hızlandırmalı paralel hesaplama stratejileri, simülasyon süresini dramatik biçimde azaltırken doğruluğu korumayı amaçlamaktadır. Bu yazıda, moleküler dinamik simülasyonlarda performansı artıran başlıca algoritmik yaklaşımların temelleri ele alınacaktır.
Klasik moleküler dinamik (MD) simülasyonlarında sistem, atomların noktasal kütleler olarak temsil edildiği ve aralarındaki etkileşimlerin kuvvet alanları aracılığıyla tanımlandığı bir çerçevede modellenmektedir. Atomlar arasındaki bağlar, açılar ve dihedraller gibi bağlı (bonded) etkileşimler ile van der Waals ve elektrostatik kuvvetleri içeren bağlı olmayan (non-bonded) etkileşimlerin toplamı, sistemin ampirik potansiyel enerji yüzeyini oluşturur (Potansiyel enerji yüzeyi ile ilgili daha fazla bilgi için tıklayın). Her bir atomun zamana bağlı konum ve hız değişimi, Newton’un ikinci hareket yasasına dayanarak hesaplanır ve bu diferansiyel denklemler sayısal integrasyon yöntemleriyle çözülmektedir. Uygulamada en yaygın kullanılan integrasyon algoritmaları arasında Verlet, Leap-Frog ve Hız-Verlet (Velocity-Verlet) yöntemleri yer almakta olup bu algoritmalar hesaplama verimliliği ile sayısal kararlılık arasında dengeli bir çözüm sunmaktadır (1–3).
Zaman integrasyon algoritmaları, sürekli zaman değişkeninin sonlu bir ızgara (grid) üzerinde ayrıklaştırılmasına dayanan sonlu farklar yaklaşımı temelinde geliştirilmiştir. Bu çerçevede zaman ekseni, ardışık noktalar arasındaki mesafenin Δt zaman adımı ile tanımlandığı ayrık zaman noktalarına bölünür (4). Newton’un hareket denklemlerinin analitik çözümü çok atomlu sistemler için mümkün olmadığından bu denklemler sayısal integrasyon yöntemleriyle yaklaşık olarak çözülmektedir. Literatürde bu amaçla geliştirilmiş çeşitli algoritmalar bulunmakla birlikte, bu yazıda moleküler dinamik simülasyonlarında en yaygın kullanılan üç temel yöntem ele alınacaktır.

Verlet Algoritması
Bu algoritmaların en basiti olan Verlet algoritması, her bir atom için mevcut konum xi(t), bir önceki zamandaki konum xi (t-Δt) ve mevcut ivme ai(t) bilgilerini kullanarak bir sonraki zaman adımındaki konumu hesaplamaktadır (5). Yöntemin matematiksel temeli, atomik konumun zamana göre ikinci dereceden Taylor açılımı ile ifade edilmesine dayanır. Bu yaklaşım, hız terimini açıkça hesaplamaya gerek kalmadan konumların güncellenmesini sağlarken ikinci mertebeden doğruluk ve iyi bir sayısal kararlılık sunmaktadır.

Denklem 1’de Δt’yi -Δt ile değiştirmek denklemde şuna yol açar:

Denklemler (Denklem 1. ve 2.) birleştirilir ve yüksek dereceli terimler atılarak aşağıdaki denklem oluşturulur:

İvmeyi tanımlayan d2r(t)/dt2 kuvvet hesabı ile elde edilebileceğinden denklemdeki, t+Δt’daki ve t-Δt’daki atomik konumlar biliniyorsa, t+Δt’daki atomik konumu döndürmelidir (Denklem 4.).

Verlet yönteminin en önemli avantajlarından biri, her bir zaman adımında kuvvet hesaplamasının yalnızca bir kez yapılmasının yeterli olmasıdır; bu durum algoritmayı hesaplama açısından verimli kılar. Ayrıca yöntem, konumların zamana göre simetrik biçimde güncellenmesine dayanması sayesinde zaman tersinirliği (time-reversibility) özelliğine sahiptir ve bu da enerji korunumu açısından sayısal kararlılık sağlar.
Bununla birlikte klasik Verlet formülasyonunda hız terimi doğrudan güncellenmez; hız genellikle konumların sonlu fark yaklaşımıyla hesaplanır. İlgili ifadede yer alan 1/Δt terimi, zaman adımı (Δt) çok küçük seçildiğinde sayısal yuvarlama hatalarının büyümesine ve hız hesaplamasında hassasiyet kaybına yol açabilir. Ayrıca konum güncellemesi hızdan bağımsız gerçekleştirildiği için hız bilgisi integrasyon şemasının doğal bir parçası değildir. Bu durum, sıcaklık kontrolü gerektiren moleküler dinamik uygulamalarında klasik Verlet algoritmasının doğrudan kullanımını sınırlamakta ve bu nedenle Leap-Frog veya Velocity-Verlet gibi hızın açıkça güncellendiği türev algoritmaların tercih edilmesine neden olmaktadır (5).

LeapFrog Algoritması
Bu sorunları aşmak için daha sonra LeapFrog yöntemi olarak adlandırılan başka bir entegrasyon algoritması geliştirilmiştir. Bu yöntemde atomik hızlar ve konumlar aşağıdaki denklemlerdeki gibi hesaplanmaktadır:

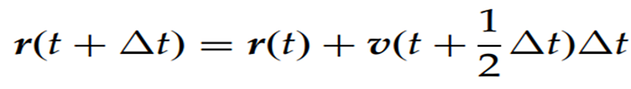
LeapFrog algoritmasında, atomlara etki eden kuvvetler ilk olarak t zamanındaki atomik konumlara göre hesaplanır ve böylece atomik ivme vektörleri a(t) elde edilir. Bir atomun t-Δt/2 anında hızı verildiğinde t+Δt/2‘deki hızı, Denklem 5.’e göre elde edilir. Daha sonra, t zamanındaki atomik hız şu şekilde hesaplanır:


Verlet yöntemiyle karşılaştırıldığında, LeapFrog yöntemi hesaplama doğruluğunu artırır ve atomik yörünge (trajectory) artık hıza bağlıdır. Böylece moleküler dinamik sisteminin harici bir termal banyo ile birleştirilmesine izin verilir. Ancak LeapFrog yöntemi, daha yüksek bilgi işlem maliyetleri ve depolama gerektirmektedir. Ayrıca hız güncellemesi her zaman konum güncellemesinden bir zaman adımı geride kalmaktadır (5).
Hız-Verlet Algoritması
Leapfrog yönteminden daha üstün bir algoritma ise Hız-Verlet yöntemi olarak adlandırılmaktadır. Bu yöntemde t + Δt/2 anındaki atomik konumlar ve hızlar, t zamanındaki değerlerinden eş zamanlı olarak elde edilir ve aşağıdaki denklemler ile hesaplanır:



Hız-Verlet algoritması, Verlet ve LeapFrog algoritmalarının ana zayıf noktasını yani hızların tanımını düzeltmektedir (6).

Sonuç olarak, zaman integrasyon algoritmaları moleküler dinamik simülasyonlarının hem doğruluğunu hem de hesaplama verimliliğini doğrudan belirleyen temel bileşenlerdir. Verlet, Leap-Frog ve Velocity-Verlet gibi yöntemler; sayısal kararlılık, enerji korunumu ve hesaplama maliyeti açısından farklı avantajlar sunmakla birlikte seçilecek algoritma simülasyonun amacı, sistem büyüklüğü ve termodinamik koşullara bağlı olarak değerlendirilmelidir. Klasik Verlet yöntemi basitliği ve zaman tersinirliği ile öne çıkarken, hızın açık biçimde güncellendiği türev algoritmalar sıcaklık kontrolü ve gelişmiş örnekleme teknikleri açısından daha esnek bir yapı sağlar. Dolayısıyla moleküler dinamikte “en iyi” algoritma tekil değildir; doğru algoritma seçimi, hız–doğruluk dengesi gözetilerek ve simülasyonun fiziksel gereksinimleri dikkate alınarak yapılmalıdır.
Referanslar
1. Cocklin, R., Heyen, J., Larry, T., Tyers, M., & Goebl, M. (2011). New insight into the role of the Cdc34 ubiquitin-conjugating enzyme in cell cycle regulation via Ace2 and Sic1. Genetics, 187(3), 701–715. https://doi.org/10.1534/genetics.110.125302
2. Papaleo, E., Casiraghi, N., Arrigoni, A. et al. (2012). Atomistic insights into the regulatory mechanisms mediated by post-translational modifications: molecular dynamics investigations. Curr. Phys. Chem. 2: 344–362.
3. Dror, R. O., Dirks, R. M., Grossman, J. P., Xu, H., & Shaw, D. E. (2012). Biomolecular simulation: a computational microscope for molecular biology. Annual review of biophysics, 41, 429–452. https://doi.org/10.1146/annurev-biophys-042910-155245
4. Verlet, L. (1967) Computer “Experiments” on Classical Fluids. I. Thermodynamical Properties of Lennard-Jones Molecules. Physical Review, 159, 98–103.https://doi.org/10.1103/PhysRev.159.98
5. Zhou K., Liu B. “Molecular Dynamic Simulation: Fundamentals and Applications” Elsevier 2022. https://doi.org/10.1016/C2017-0-04711-0
6. Boisseau P., Houdy P., Lahmani M. “Nanoscience Nanobiotechnology and Nanobiology” Springer-Verlag Berlin Heidelberg 2009. https://doi.org/10.1007/978-3-540-88633-4



Yorumlar