
PCR: A Revolutionary Technique in Molecular Biology
- Muhammed Furkan Üstün

- Jul 9, 2025
- 7 min read
With the incredible advancements in biology and molecular genetics over the past century, a significant amount of genetic data has been generated and accumulated. Alongside technological progress, this large volume of genetic information has transformed into an interdisciplinary field—bioinformatics—that not only enables the processing and storage of these data but also makes them accessible for various users (1). For example, in 1986, the average number of genomic data entries (nucleotide sequences) was around 9,980, whereas in 2001 this number rose to 14,976,310, and today it has reached approximately 4.7 billion, continuing to grow rapidly (2,3). If you would like to explore more on this topic, we recommend checking out our article titled “Introduction to Bioinformatics and Basic Concepts”. Dear science enthusiasts, in this article, we will focus on one of the foundational tools that played a crucial role in the emergence of bioinformatics: PCR (Polymerase Chain Reaction).
Polymerase Chain Reaction (PCR)
PCR is an in vitro biotechnological technique used to enzymatically amplify and replicate a specific DNA region located between two known loci. It consists of three main stages: denaturation, annealing, and extension. The method was initially developed and applied by Kjell Kleppe. However, during that time, the technique had several limitations due to the frequent use of materials and high error rates. One of the most critical issues was that the DNA polymerase enzyme—which extends the DNA strand by adding nucleotides to its complement—would denature when exposed to the high temperatures (90–93°C) required in the “Denaturation” phase of each cycle. As a result, the enzyme had to be re-added after every cycle, significantly increasing the error rate and reducing efficiency.
This challenge was overcome in 1985 when Kary Mullis introduced the use of Taq polymerase, an enzyme isolated from heat-resistant archaea (Thermus aquaticus). Taq polymerase could withstand high temperatures and function effectively during the annealing and extension steps, making the PCR process much more reliable. This breakthrough led to the development of what we now know as Polymerase Chain Reaction (PCR) technology and earned Kary Mullis the 1993 Nobel Prize in Chemistry (4).

PCR is a serial technique used for DNA cloning, and as such, it has enhanced the power of recombinant DNA research by eliminating the need for host cells in the DNA cloning process. PCR enables the amplification of a target DNA sequence—even one that is present in extremely low amounts—within a population or sample containing various DNA molecules, through a series of in vitro reactions. With each cycle, the amount of DNA theoretically doubles (2ⁿ amplification).
However, in order for PCR to proceed accurately and efficiently, specific components must be present and properly used. These essential materials include:
DNA Template
Primers (two are required: forward and reverse)
dNTPs (dATP, dTTP, dGTP, dCTP)
Taq DNA Polymerase
Buffer and Ion Solutions
Nuclease- and DNase-Free Water
To amplify the desired specific region, primers—which are short oligonucleotide sequences identifying the target locus—must be carefully designed. Primers are typically 18–24 nucleotides in length and are designed to recognize the 5’-3’ and 3’-5’ ends of the region to be amplified. During the PCR cycle, these primers anneal to the complementary sequences on the DNA template and allow the extension of the remaining strand.
Each complete cycle that successfully doubles the amount of target DNA is referred to as one PCR cycle. In this way, the quantity of DNA increases exponentially—doubling with each cycle—and within just a few hours, a substantial amount of DNA can be obtained from a minimal starting material (4).

The amount of amplified DNA depends on the number of systematically repeated cycles and the materials present in the reaction tube. However, several factors can hinder DNA amplification or the generation of error-free sequences. Common PCR protocols typically consist of three main cycles, and it is evident that any error occurring in one of these steps can negatively affect the subsequent reactions. These three steps are as follows:
Denaturation
The double-stranded DNA selected for cloning must be converted into single strands to allow primers to bind and extend. Therefore, the DNA is denatured. This is achieved by heating the double-stranded DNA to approximately 92–95°C for about 1 minute, which separates the strands into single-stranded DNA (4).
Annealing (Hybridization)
Following denaturation, the DNA exists in a single-stranded form. The reaction temperature is then lowered to between 45–65°C, allowing the pre-designed primers, which correspond to the target region, to bind to their complementary sequences on the DNA strands. Once primers are in place, Taq polymerase uses them as starting points during the extension step. This phase is referred to as annealing or hybridization (4).
Extension (Elongation)
After annealing, Taq polymerase must extend the primers to replicate the desired region. For this, the temperature is adjusted to between 65–75°C. During this step, the enzyme sequentially adds complementary nucleotides to the growing DNA strand from the 5’ end toward the 3’ end, thus synthesizing a new copy of the target DNA sequence (4).

As seen in Figure 3, the presence of Van Cat cDNA obtained after PCR is demonstrated through an Agarose Gel Electrophoresis application. The bright bands within the marked area indicate that the DNA migrated through the gel in response to the applied electric current. However, since the cDNA in this case was not amplified using any primers and possesses a large sequence, it remained mostly intact and unamplified. As a result, its movement within the gel was limited under the given current.
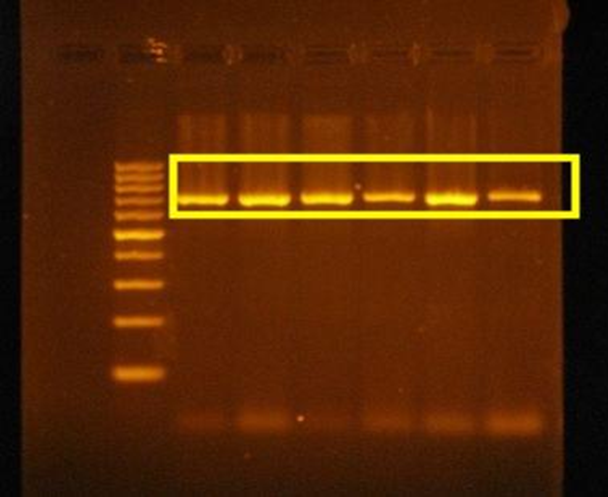
As seen in Figure 4, the PCR analysis of the Van Cat genome using primers has been demonstrated. In this study, the pre-designed primers for the target gene region bind to the gene regions and are amplified through PCR, followed by Agarose Gel Electrophoresis to confirm the specificity of the amplified region. The areas within the yellow zone in the results indicate that the primers successfully bound to the corresponding gene regions, and after amplification, the gene size increased.
Since the discovery of PCR, many new methods have emerged. Although these methods differ from each other, the primary goal has always been to produce specific copies of the desired region. These methods include Nested-PCR, Real-Time PCR, Multiplex PCR, Reverse PCR, and Touchdown PCR.
Nested-PCR
This method aims to increase the specificity of PCR, i.e., to amplify the desired region accurately and without error. It involves a two-step PCR technique. First, a sequence outside the target region is selected for amplification. Then, within the amplified outer region, the actual sequence to be amplified is chosen, and PCR is performed again to obtain the target region. In this method, two PCRs are required, so primers for the larger, outer sequence are designed first. Then, specific primers for the inner region to be amplified are designed, and PCR is performed. This method allows for more specific and error-free amplification of the target region (6).
Real-Time PCR
This method provides researchers with real-time data on the quantity of PCR products during the experiment. Fluorescent signals are sent to the tube cyclically, and the level of amplification of the target region is analyzed and presented simultaneously. Unlike standard PCR, where the results can only be measured after the PCR process using gel electrophoresis, Real-Time PCR allows measurement of DNA amplification at each cycle using fluorescent signals (4).
Multiplex PCR
This method is used to amplify the entire genomic DNA in PCR. Multiple primer pairs targeting different regions along the DNA sequence are designed and used to amplify multiple regions of the genome simultaneously. This method can be thought of as producing "Okazaki fragments." It is particularly useful for rapidly amplifying short sequences from viruses and bacteria, allowing for quick identification (7,8).
Reverse Transcription PCR
Unlike standard PCR, this method utilizes the principle of RNA replication in retroviruses, which have RNA as their genomic material. Using the Reverse Transcriptase enzyme isolated from these viruses, complementary DNA (cDNA) is synthesized from RNA. This method allows for the easy and rapid analysis of gene expression. Using the enzyme isolated from retroviruses, RNA is used as a template to synthesize DNA specific to the target genome. After the DNA-RNA hybrid is separated, the resulting DNA sequence is used to generate double-stranded DNA, which is then amplified (9).
Touchdown PCR
Touchdown PCR (TD-PCR) is a technique developed to increase the specificity of PCR compared to the classic PCR protocol. It is especially preferred for ensuring that primers bind specifically to the target DNA. In this method, the annealing temperature used during PCR is initially raised above the normal temperature for the first cycle and is then gradually reduced over several cycles. This temperature ensures that primers can only bind to fully complementary sequences. Subsequently, the temperature is decreased by 1-2°C every few cycles to an optimal temperature where primers can bind with less stringent matching. This approach ensures high specificity in the initial cycles and increases amplification efficiency in subsequent cycles, allowing for more primer binding. It helps prevent the binding of incorrect primers and ensures the binding of high-specificity primers, resulting in stronger PCR products (4).
As a revolutionary discovery in the field of molecular biology and genetics, Polymerase Chain Reaction (PCR) has been continuously evolving since its discovery and finding its place in various application areas. Thanks to the development of new methods and protocols every day, PCR is now widely used across a broad spectrum, from diagnostics to forensic medicine, from agriculture to evolutionary biology. Efforts to make this technology more precise, faster, and specific continue to make significant contributions to the scientific community. With the strong infrastructure provided by PCR, it is expected that genetic analyses will become much more accessible, reliable, and cost-effective in the future. In this context, understanding the foundations of PCR technology and its evolving variants provides invaluable knowledge for current and future research.
References
1. Koyun, H., ve Üstün, M. F., (2024). Bioinformatic Comparisons of Some Web-based PCR Primer Design Programs. Hayvan Bilimi ve Ürünleri Dergisi. 7(2), 134-144. https://doi.org/10.51970/jasp.1596993
2. National Library of Medicine (NCBI). https://www.ncbi.nlm.nih.gov.
Erişim Tarihi: 28.06.2025.
3. Atalay, R. Ç., (2002). Neden Biyoinformatik. Avrasya Dosyası, Moleküler Biyoloji ve Gen Teknolojileri Özel, Sonbahar. 8(3), 129-141.
4. Klug, W. S., Cummings, M. R., Spencer, C. A., ve Öner, C., (2009). Genetik kavramlar. Palme Yayıncılık, p. 500-512.
6. Karataş M. Moleküler Biyoloji Nobel Akademik Yayıncılık. (2012), p. 288-290.
7. Colinet, D, J., Kummert, P. Lepoivre and J. Semal., 1994. Identification of distinct potyviruses in mixedly- infected sweetpotato by the polymerase chain reaction with degenerate primers. Phytopathology. 84, 65-69.
8. Levy, L., A. Hadidi and Garsey, S. M. (1992). Reverse transcription-polymerase chain reaction asays for the rapid detection of citrus viroids using multiplex primer sets. Proc. Int. Soc. Citriculture, 2, 800.
9. Santagati, S., Garnier, M., Carlo, P., Violani, E., Picotti, G, B., Maggi, A., (1997). Quantitation of low abundance mRNAs in glial cells using different polymerase chain reaction (PCR)-based methods, Br Res Prot; 217.



Comments