
AutoDock Vina ve ChimeraX ile Moleküler Yerleştirme (Docking) İş Akışı
- Yağmur Çavaş

- 8 Eki
- 5 dakikada okunur
Moleküler yerleştirme (docking), bir ligandın hedef proteine ait bağlanma bölgesine nasıl ve ne kadar güçlü bir şekilde bağlandığını tahmin etmek için kullanılan bir in silico yöntemdir. İlaç keşfi ve ilaç tasarımında yaygın olarak kullanılmaktadır (1). Bu yazı, UCSF ChimeraX kullanarak temel moleküler yerleştirmeye hazırlık adımlarını ve M1 MacBook Air üzerinde Autodock Vina ile yerleştirme (docking) işlemini açıklamaktadır.
Yerleştirme Prosedürü için Gereksinimler
Moleküler yerleştirme, Windows 10 veya üstü, macOS (10.15 Catalina veya üstü) ve Linux dahil olmak üzere tüm büyük işletim sistemleriyle uyumlu olan AutoDock Vina kullanılarak gerçekleştirilebilir. Moleküler yapı hazırlama ve görselleştirme için UCSF Chimera veya ChimeraX (sürüm 1.8 veya üstü) gibi yardımcı yazılımlar kullanılabilir (2-4).
1. Adım: Bu rehberde, protein hedefi olarak COVID-19 ana proteazının inhibitör N3 ile kompleks halindeki kristal yapısı (PDB ID: 6LU7) kullanılmıştır. Hedef protein, Protein Veri Bankası'ndan (PDB) alındıktan sonra, Şekil 1.'de görüldüğü gibi PDB formatında kaydedilir ve ChimeraX'te açılır.

2. Adım: Aktif bölgeyi doğru bir şekilde tanımlamak için, mevcut inhibitörü tanımlamak çok önemlidir. Bu amaçla, Chimera menüsünde yer alan Select > Chains > N-[(5-METİLİSOXAZOL-3-İL)KARBONİL]ALANİL-L-VALİL-N~1~-((1R,2Z)-4-(BENZİLOKSİ)-4-OKSO-1-{[(3R)-2-OKSOPİROLİDİN-3-İL] METHYL}BUT-2-ENYL)-LLEUCINAMIDE seçeneğine tıklanarak inhibitör seçilir.
İnhibitör seçildikten sonra zincirler renklendirilmelidir. Seçilen kısımları proteinin geri kalanından ayırt etmek için (Şekil 3.), Eylemler > Renk > mor (veya tercih ettiğiniz herhangi bir renk) seçeneğine giderek renk değiştirilir.

3. Adım: Ardından, moleküler yerleştirme için proteini optimize etmek üzere Dock Prep araçları kullanılır. Bunun için, Chimera menüsündeki Tools > Structure Editing > Dock Prep (Şekil 4.) seçeneğine tıklanır.
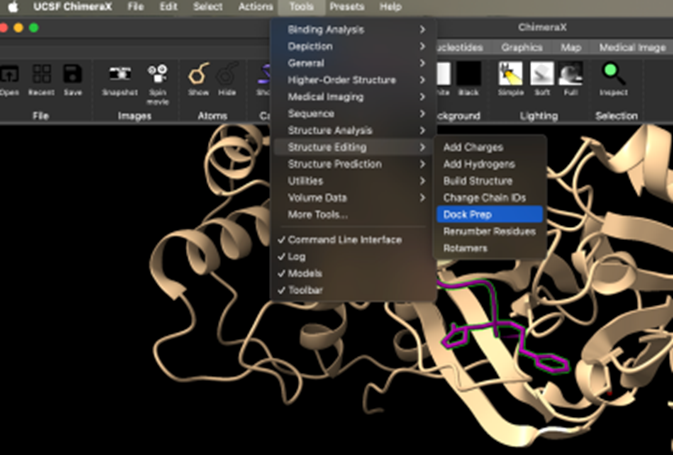
Açılan sekmede “Delete non-complexed ions” seçeneği hariç, Dock Prep kutusundaki tüm seçenekler seçilir ve ardından Ok'a tıklanır (Şekil 5.).
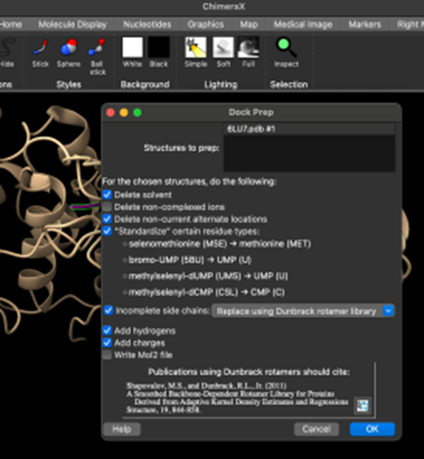
4. Adım: Ardından, aşağıdaki ilgili parametreler seçilip Ok'a tıklanarak proteine hidrojenler eklenir (Şekil 6.). Yazılımın, daha önce bahsedilen alternatifleri seçerek modele göre en uygun seçeneği seçmesine izin verilmektedir.
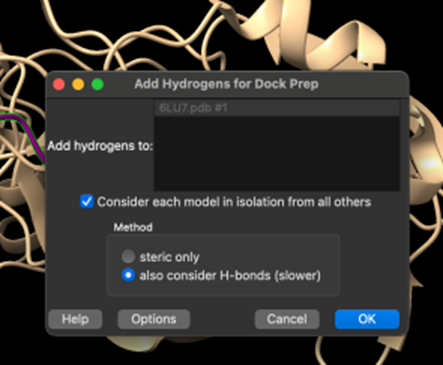
Şekil 7.’de verildiği üzere, proteine Gasteiger metodu kullanılarak uygun yükler eklenir ve Ok’a basılır.

5. Adım: Ardından, PDB yapısındaki ligandlar (bu durumda önceden renklendirilmiş olan Zincir C) silinir ve bu dosya pre_6lu7.pdb (istenilen isimde) olarak kaydedilir (5).

Moleküler Yerleştirme (Docking)
Moleküler yerleştirmede, potansiyel bağlanma pozisyonlarının aranması bu tanımlanmış bölgeyle sınırlı olduğundan, ızgara kutusunun (grid box) tanımı kritik bir adımdır. Izgara kutusunun boyutu ve koordinatları birkaç yaklaşımla belirlenebilir:
1. Literatür Verilerine Dayalı
Daha önce yayınlanmış yerleştirme çalışmalarında genellikle belirli protein-ligand sistemleri için optimize edilmiş ızgara parametreleri bildirilir. Bu değerlerin kullanılması, çalışmalar arasında sonuçların karşılaştırılabilirliğini ve tekrarlanabilirliğini sağlamaktadır.
2. Ko-Kristalize (Doğal) Ligand Kullanımı
Protein yapısı ko-kristalize bir ligand içeriyorsa bu ligandın geometrik merkezi, ızgara kutusu merkezini tanımlamak için kullanılabilir. Bu yöntem, yerleştirmenin deneysel olarak doğrulanmış bağlanma bölgesine odaklanmasını sağlamaktadır. ChimeraX veya PyMOL gibi moleküler görselleştirme araçları, ligandın merkez koordinatlarını hesaplamak için kullanılabilir.
3. Aktif Bölge Rezidüleri ile Tanımlama
Anahtar katalitik veya bağlanma rezidüleri biliniyorsa (örneğin, SARS-CoV-2 ana proteazındaki His41 ve Cys145), kutu bu rezidülerin koordinatlarının geometrik ortalamasına göre ortalanabilir. Kutunun boyutları, genellikle 20 ila 30 Å arasında değişen beklenen ligand boyutuna göre ayarlanmalıdır.
4. Kör Bağlanma Yaklaşımı (Blind Docking)
Bağlanma bölgesi bilgisi bulunmayan durumlarda, tüm proteini kapsayan yeterince büyük bir ızgara kutusu uygulanabilir. Bu strateji, potansiyel bağlanma boşluklarının tarafsız bir şekilde araştırılmasını sağlar ancak hesaplama açısından daha zordur (hesaplama süresi nispeten daha uzundur).
Bu rehberde, ızgara kutusu koordinatları literatürden alınmıştır (6,7). Ancak buna alternatif olarak ko-kristalize ligandın merkez noktası, aktif bölge koordinatları veya tüm proteini kapsayan kör yerleştirme (blind docking) stratejisi kullanılarak hesaplanabilir.
1. Adım: AutoDock Vina'yı sisteminize başarıyla yükledikten sonra, yerleştirme işlemini gerçekleştirmek için PDBQT dosyalarının oluşturulması gerekmektedir. Bu adımda, polar hidrojen atomlarını ve bunlara karşılık gelen kısmi yükleri içeren hedef proteinin PDBQT dosyasını oluşturulacaktır. Bu aşama için terminalde mk_prepare_receptor.py dosyası kullanılacaktır.
$ mk_prepare_receptor.py -i prep_6lu7.pdb -o 6lu7_receptor -p -v --box_size 25 25 25 --box_center -11.824 14.735 74.152
Bu komutta, -i komutu girdi (input) protein yapısını (prep_6lu7.pdb) belirtirken -o komutu çıktı (output) ön ekini (6lu7_receptor) tanımlar. -p ve -v seçenekleri sırasıyla protonlanma ve ayrıntılı çıktıyı etkinleştirir. Yerleştirme için arama alanı (search area) --box_size 25 25 25 ile tanımlanmıştır ve --box_center -11.824 14.735 74.152 koordinatlarını merkez alan, kenarları 25 Å olan kübik bir kutuyu belirtir.
2. Adım: Bu işlemin ardından üç temel çıktı dosyası oluşturulur:
1. 6lu7_receptor.pdbqt – AutoDock Vina için gerekli olan rijit proteine ait girdi dosyası.
2. 6lu7_receptor.box.txt – Vina formatında kutu boyutlarını içeren bir metin dosyası.
3. 6lu7_receptor.box.pdb – tanımlanan ızgara kutusunun görselleştirilmesini sağlayan bir PDB
dosyası.
Benzer şekilde, Meeko python paketi kullanılarak liganda ait bir PDBQT dosyasının da oluşturulması gerekiyor.
3. Adım: Ligandın uygun şekilde hazırlanması, moleküler yerleştirmeden önce kritik bir adımdır. Çünkü molekülün yerleştirme yazılımı için uygun bir formatta ve konformasyonda olmasını sağlamaktadır. Bu çalışmada, quercetin molekülü AutoDock Vina ile uyumlu olarak .pdbqt formatına dönüştürülmüştür. Prosedür şu şekildedir:
Quercetin'in üç boyutlu yapısı PubChem veri tabanından .sdf formatında elde edilir. mk_prepare_ligand.py kodu liganda ait .pdbqt dosyasının oluşturulması için kullanılır.
$ mk_prepare_ligand.py -i Conformer3D_COMPOUND_CID_5280343.sdf -o quercetin.pdbqt
4. Adım: Bu adımın ardından quercetin, moleküler yerleştirme için AutoDock Vina ile tamamen uyumlu bir .pdbqt dosyası olarak başarıyla hazırlanmıştır. Bu, ligandın doğru bağlanma tahminleri için uygun protonlanma durumlarına, döndürülebilir bağlara ve atomik yüklere sahip olmasını sağlamaktadır.
Artık aşağıdaki komut çalıştırılarak moleküler yerleştirme analizi başlatılabilir.
$ vina --receptor 6lu7_receptor.pdbqt --ligand quercetin.pdbqt --config 6lu7_receptor.box.txt
--exhaustiveness=32 --out 6lu7_ligand_vina_out.pdbqt
Exhaustiveness (Kapsamlılık) parametresi 32 olarak ayarlandığında elde edilen bulgular, genellikle bu enerji seviyesinde tek bir yerleştirilmiş (docked) konformasyon üretmektedir. Varsayılan kapsamlılığın azaltılması nedeniyle, birkaç poz ters sırayla sunulabilir ve daha az uygun enerji sergileyebilir.


Referanslar
1.Butt, S. S., Badshah, Y., Shabbir, M., & Rafiq, M. (2020). Molecular Docking Using Chimera and Autodock Vina Software for Nonbioinformaticians. JMIR bioinformatics and biotechnology, 1(1), e14232. https://doi.org/10.2196/14232
2.Eberhardt, J., Santos-Martins, D., Tillack, A. F., & Forli, S. (2021). AutoDock Vina 1.2.0: New Docking Methods, Expanded Force Field, and Python Bindings. Journal of chemical information and modeling, 61(8), 3891–3898. https://doi.org/10.1021/acs.jcim.1c00203
3.Trott, O., & Olson, A. J. (2010). AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. Journal of computational chemistry, 31(2), 455–461. https://doi.org/10.1002/jcc.21334
4.Meng, E. C., Goddard, T. D., Pettersen, E. F., Couch, G. S., Pearson, Z. J., Morris, J. H., & Ferrin, T. E. (2023). UCSF ChimeraX: Tools for structure building and analysis. Protein science : a publication of the Protein Society, 32(11), e4792. https://doi.org/10.1002/pro.4792
5.Bilginer, S., Gözcü, S., & Güvenalp, Z. (2022). Molecular Docking Study of Several Seconder Metabolites from Medicinal Plants as Potential Inhibitors of COVID-19 Main Protease. Turkish journal of pharmaceutical sciences, 19(4), 431–441. https://doi.org/10.4274/tjps.galenos.2021.83548
6.Yahya, E. M. (2020). Criblage des molécules inhibitrices du SARS-CoV-2 par une approche in silico.
7.Junapudi, S., Janapati, Y. K., Uppugalla, S., Harris, T., Yaseen, M., & Latif, M. (2023). Molecular Docking Analysis of SARS-CoV-2 Inhibitor N3 (6LU7) against Selected Flavonoids and Vitamins. Coronaviruses, 4(4), 29-36. https://doi.org/10.2174/0126667975261384231010181117



Yorumlar